
Click now to listen to RCA’s Director of Regulatory Affairs, Jordan Elder, in this RCA Radio audio update:
The European Parliament recently voted for a timeline extension to MDR regulation, including an extended timeline for medical device regulatory submission. The 537-3 vote by members of the official body provides a final approval to extend MDR deadlines for compliance. Included in the legislation is revisions to regulatory submission rules for certifying medical devices. Additionally, the timeline includes new dates for both Regulation (EU) 2017/745 (MDR) and Regulation (EU) 2017/746 (IVDR).
Regulated Companies
European officials extended the MDR timeline to include legacy devices with existing certificates under the MDD to stay on the market until 2024. Industry executives have suspected for some time that the EU medical device backlog of submissions would lead to this type of scenario. Equally important, many EU health ministers voiced their opinion during a December 2022 session medical device shortages would occur without a MDR timeline extension.
MDR Timeline
Manufacturers now have until 2027 or 2028 to have medical devices certified and approved under MDR guidelines. The timeline includes new dates for both high-risk medical device products and low risk medical devices. For medical devices covered by a certificate or a declaration of conformity issued before 26 May 2021, the transition period to the new rules is extended from 26 May 2024 to:
- 31 December 2027 for Class III and IIb;
- 31 December 2028 for Class IIa and I.
- 26 May 2026 for Class III implantable custom-made devices
The original “sell-off’ date requirement that was required under the MDR has been removed
Need help with your MDR or IVDR transition? Talk to our Experts →
IVDR Timeline
Meanwhile, the EU Commission has previously recommended the IVDR application date be extended due to the EU notified body bottleneck.
- General EU MDR Class 1 Low-risk devices that are non-measuring, non-sterile, non-reusable, non-surgical, and that do not require review from a notified body will still go into effect in 2022.
- Non-sterile Class A and B Devices (low risk) – May 26, 2022
- Class D (Highest Risk) – May 26, 2025
- Class C (Medium Risk) – May 26, 2026
- Sterile Class A and B Devices (low risk) – May 26, 2027
Industry Reaction
Life science media outlets have reported extensively on the change since it was proposed at the meeting.
Stella Kyriakides, the European health commissioner, first proposed postponing the current MDR deadline dates during the EPSCO council meeting in Brussels. The health commissioner projected around 23,000 devices and 1,500 IVDs are currently approved and certified under MDD, but have not yet transitioned to the new MDR regulation. These medical devices are likely to expire in 2024 and 2025,
“The transition to the new rules has been slower than we anticipated,” said Kyriakides. “The pandemic, shortages of raw materials caused by the Russian invasion against Ukraine and low notified body capacity has put a strain on market readiness.”
Medical Device Manufacturers
Additionally, many unique viewpoints have emerged about the current landscape and the impact of recertification.
“If the three-year deadline is truly unattainable, these extensions could prevent devices that are perfectly safe for use from being taken off the market because they were unable to get recertified in time,” said Alexandra Murdoch, a medical analyst at GlobalData.
Murdoch added both medical device manufacturers and suppliers must now deliver MDR regulatory documentation for market approval, including data about materials used in medical device manufacturing.
Medical Device Safety
Both Pinto & Rocha have documented the MDR proposal concerns that “only medical devices considered safe will benefit from the extension”. This includes medical device manufacturers that have begun the process of submission and certification under the MDR.
Further, the Commission has recognized the ongoing need for patient safety and proposed a 2023 pilot project for medical device manufacturers. For example, expert panels to advise manufacturers with qualified scientific advice about devices that help treat rare diseases.
Medical Device Shortage
Ireland’s minister of health, Stephen Donnelly, supported the change based on COVID-19 procurement and preventing future medical device shortage scenarios.
“Participation in the EU COVID-19 vaccine strategy has allowed us to conduct the largest immunization program in our country’s history, saving countless lives and enabling the resumption of normal social and economic life.” said Donnelly.
“This measure needs to be adopted and take effect without delay to ensure that the devices our citizens and health systems rely on remain available.”
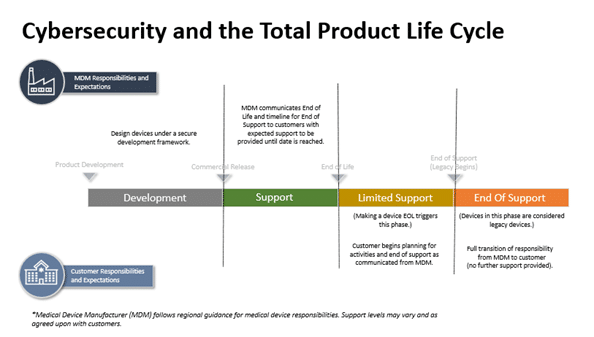
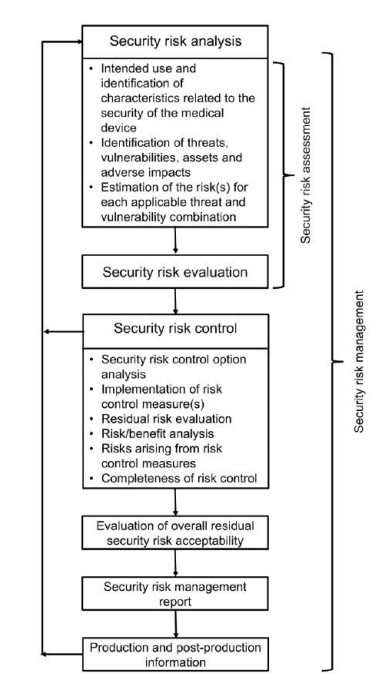
Cybersecurity
By contrast, it is still unknown how different types of EU legislation will regulate connected medical devices. There is concern across the industry about which legislation may take precedence and the level of postmarket surveillance data needed.
The European Commission (EC) published a proposal for a Cyber Resilience Act (“CRA”) to strengthen cybersecurity across medical device interoperability. Both Wright & Wenzel have documented this legislation does not consider MDR to impose as many obligations on medical device manufacturers. Further, the commentary suggests the EC may not require as much documentation about unknown vulnerabilities are not present for medical devices.
About RCA’s Medical Device Consulting Services
The regulatory compliance process surrounding the medical device industry involves a strict adherence to pre/post market information throughout a device’s life cycle. Even a single compliance issue you have can turn into a significant effect on your business. Regulatory Compliance Associates can help guide you through any stage of the medical device consulting process, with capabilities during product development through the regulatory clearance/approval of your product.
Our team of over 500 medical device consulting Experts — including former FDA officials and regulatory compliance leaders in the field of medical device regulation — will work with your company to create a quality assurance and regulatory compliance approach tailored to your products and regulatory needs. Regulatory Compliance Associates works with international Fortune 100 companies, venture capital start ups, and companies of all sizes and shapes. our compliance enforcement solutions for law firms include remediation for warning letters, FDA 483’s, import bans or consent decrees. Very few regulatory compliance services have the same regulatory compliance expertise in a variety of medical fields.
Cybersecurity
For medical device manufacturers, technology can be a double-edged sword. The innovative technologies that elevate the quality of life for patients can also be used to potentially undermine the organization using the device. The consequences can affect the device itself if Regulatory Compliance Associates medtech consultants do not implement good IoT cybersecurity and FDA cybersecurity protocols.
At Regulatory Compliance Associates, we offer a wide variety of services for medical devices security to help ensure that your product is protected from cyber-attacks. With a well-planned design, along with full visibility of product development and the supply chain, Regulatory Compliance Associates medical device consultant Experts can help strengthen your device’s cybersecurity. We partner with medical device companies in each phase of the design cycle, including protecting inputs from threat exposure and hardening outputs for regulatory compliance & FDA submission approval of your medical technology.
- SaMD Consulting
- Threat Modeling
- Proof of Concept
- Quality Assurance Services
- TIR 57 & TIR 97
- ISO 62304
- ISO 27001
Regulatory Affairs
Regulatory affairs is Regulatory Compliance Associates® backbone, and we handle more submissions in a month than many manufacturers do in a lifetime. Our regulatory compliance consulting Experts have experience working with the FDA, global regulatory bodies and / or agencies, and notified bodies worldwide. Therefore, you can count on us for in-depth and up-to-date insights which increase speed-to-market.
As a trusted regulatory affairs consultant, our FDA veterans and industry experts represent Regulatory Compliance Associates® as one of the top medical device consulting firms. We’re here to help you navigate the difficulties associated with new product submissions. Regulatory Compliance Associates® medical device consulting company has expertise in both the approval process and post-approval support.
- New Product Approval
- Post-Approval Support
- Outsourced Staffing
- EU MDR
- Combination Products
Compliance Assurance
Increasingly, life science companies are feeling the pressure of greater scrutiny by regulators, and responding by developing sustainable compliance strategies. Whether it’s preparing for an audit, developing a response to an FDA finding, or remediation to an adverse event, Regulatory Compliance Associates® can help.
Our network of over 500 medical device consultant & FDA, MHRA & EMA veterans are industry professionals offers a unique blend of expertise. This allows Regulatory Compliance Associates® to handle both simple and complex regulatory compliance challenges within medical device consulting companies.
- Gap Assessments
- Internal Audits
- Employee Training
- Notified Body Response
- Data Integrity
Quality Assurance
Regulatory Compliance Associates® Quality Assurance consulting includes quality system assessments, strategy, implementations, and identification of quality metrics to ensure continuous improvement, aligning with your business needs and goals. Each Regulatory Compliance Associates® medical device consultant is a quality expert with experience spanning major corporations and start-ups. We know firsthand how to achieve, maintain, and improve quality, and we excel in transferring this knowledge to your organization.
In the medical devices field, quality assurance (QA) is more than merely ensuring the quality of a finished product. You need the tools to monitor and regulate every process from the design of a new product to continued quality compliance as the device is sent to market. At Regulatory Compliance Associates®, we offer you the quality assurance services you need to monitor these processes and ensure quality compliance every step of the way.
With more than 20 years experience working with medical device consulting companies, Regulatory Compliance Associates® trusted medical device quality assurance consultant team is fully equipped to handle your unique QA needs.
- ISO13485
- 21 CFR 210
- 21 CFR 211
- Outsourced Staffing
- MDSAP
- Facility Validation
- Equipment Validation
- Quality Metrics
Remediation Services
Regulatory Compliance Associates® is widely recognized within medical device consulting companies & the life science industry for our remediation services & support. Regulatory Compliance Associates® ability to help companies successfully resolve complex regulatory challenges have a proven track record of success. Our medical device consulting services include significant experience with the development of responses to 483 Observations, Warning Letters, Untitled Letters and Consent Decrees.
- Regulatory Action
- Regulatory Compliance
- Regulatory Enforcement
- Warning Letter
- 483 Observation
- Oversight Services
Our value goes beyond the initial response by helping companies successfully execute their action plans, develop an improved compliance culture tailored to the needs of their business, and ultimately move beyond the regulatory action to emerge as a stronger business. We negotiate difficult demands of remediation with insight and the clear advantage of our medical device consultant expertise and experience that makes partnering with Regulatory Compliance Associates® a competitive differentiator in the remediation space.
- Quality System
- Technical File
- Design History File
- Data Integrity
- cGMP
Strategic Consulting
Whether it’s a strategy, a technical plan, or project, Regulatory Compliance Associates® medical device consultancy can help ensure a successful project. Regulatory Compliance Associates® medical device strategy consulting can deliver your project on time, on budget, and you’re never embroiled in a costly mistake.
Our medical device consultant Experts are industry Experts are here to provide the unique insight you need before an M&A deal, through a staffing crisis and in every area of your product’s development and life cycle. As the trusted medical device manufacturing consultants of thousands of companies around the world, we have the knowledge and expertise needed to deliver exceptional results to your business — no matter your size or unique needs.
- Manufacturing Optimization
- Product Lifecycle Management
- Mergers & Acquisitions (M&A)
- Due Diligence
- Device Vigilance
- Risk Management Plan
- Product Complaints
- Medical Information
About Regulatory Compliance Associates
 Regulatory Compliance Associates® (RCA) provides medical device consulting to the following industries for resolution of life science challenges:
Regulatory Compliance Associates® (RCA) provides medical device consulting to the following industries for resolution of life science challenges:
- Life Sciences
- Pharmaceutical
- Biologic & Biotechnology
- Sterile compounding
- Medical device
- Lab Testing
We understand the complexities of running a life science business and possess areas of expertise that include every facet of R&D, operations, regulatory affairs, quality, and manufacturing. We are used to working on the front lines and thriving in the scrutiny of FDA, Health Canada, MHRA and globally-regulated companies.
As your partners, Regulatory Compliance Associates can negotiate the potential minefield of regulatory compliance and regulatory due diligence with insight, hindsight, and the clear advantage of our unique expertise and experience.
- Founded in 2000
- Headquartered in Wisconsin (USA)
- Expertise backed by over 500 industry subject matter experts
- Acquired by Sotera Health in 2021
About Sotera Health
The name Sotera Health was inspired by Soteria, the Greek goddess of safety, and reflects the Company’s unwavering commitment to its mission, Safeguarding Global Health®.
Sotera Health Company, along with its three best-in-class businesses – Sterigenics®, Nordion® and Nelson Labs®, is a leading global provider of mission-critical end-to-end sterilization solutions and lab testing and advisory services for the healthcare industry. With a combined tenure across our businesses of nearly 200 years and our industry-recognized scientific and technological expertise, we help to ensure the safety of over 190 million patients and healthcare practitioners around the world every year.
We are a trusted partner to more than 5,800 customers in over 50 countries, including 40 of the top 50 medical device companies and 8 of the top 10 pharmaceutical companies.
Commitment to Quality
Our Certificate of Registration demonstrates that our Quality Management System meets the requirements of ISO 9001:2015, an internationally recognized standard of quality.
To begin the Regulatory Compliance Associates® scoping process today, please enter your information in the blue form below and click the submit button at the bottom of the webpage.


 Regulatory Compliance Associates (
Regulatory Compliance Associates (