The International Medical Device Regulation Forum (IMDRF) recently published updated cybersecurity guidance for the medical device industry. The medical device cybersecurity working groups at IMDRF have been busy lately, publishing multiple final documents about medical devices & software as medical device (SaMD).
Regulatory Compliance
IMDRF’s medical device guidance provides steering assumptions for both regulatory compliance & medical device cybersecurity, which are appropriate for sponsors developing medical devices. Further, a primary objective of the guidance is simultaneously increasing patient safety & reducing external threats for providers and HCPs.
Global Harmonization
The guidance begins with harmonization concepts that could affect multiple departments inside a medical device manufacturer. Additionally, key areas for harmonization programs highlighted by the cybersecurity guidance include:
- Product design
- Risk management activities
- Device labelling
- Regulatory submission
- Information sharing
- Post-market activities
Product Life Cycle (PLC)
IMDRF’s cybersecurity guidance continues on with a deeper evaluation of risks associated across the product life cycle. It is recommended for potential vulnerabilities to be considered for any product life cycle stage, especially considering legacy devices that may be vulnerable to strategic risk.
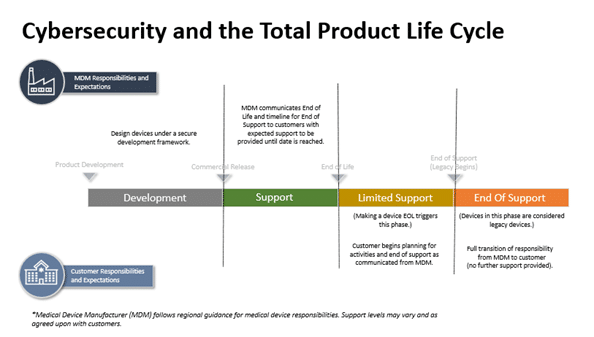
Product Design
Product design considerations include the initial phases of medical device development and continues until the end of support (EOS) once a product is discontinued. The four product design stages the cybersecurity guidance refers to when it comes to total product life cycle:
- Development Stage
- Support Stage
- Limited Stage
- End of Support
Development Stage (Stage 1)
The Development Stage occurs during the pre-commercialization phase before a medical device is approved by a regulatory body. This is when medical device manufacturers begin to incorporate security into the product concepts being designed. Design controls are critical in this stage for medical device manufacturers to leverage when considering how to mitigate risks.
Finally, an important deliverable of the Development Stage is product-related security documentation. The documentation is designed to help unfamiliar users to understand how to securely operate the medical device.
Support Stage (Stage 2)
The Support stage is during the initial post-launch phase and may continue for many years. Medical devices in this stage are:
- Currently used for providing patient care
- Available for purchase on the open market
- Contain major software, firmware, or programmable hardware components
- Support for software, firmware or components is provided by the medical device manufacturer
Additionally, medical devices in the Support stage should receive full cybersecurity support. This support often includes software patches, software updates, hardware updates, and incremental support the manufacturer considers appropriate.
Limited Support Stage (Stage 3)
Medical device manufacturers continue to provide cybersecurity support during Stage 3. However, as product development transitions to a more current medical device design, different constraints are involved with the transition. Medical devices in Stage 3 often require additional network controls compared to medical devices in Stage 2:
- Third-party components or software may be used more frequently than internally developed updates or patches
- Cybersecurity best practices integration is often governed by the ease of following support practices outlined in the Stage 2
- Medical device manufacturers must explain to users the existing limitations that are now recognized in the devices and services affected
- Healthcare providers using the medical device should begin to take more of an active role in unmitigated features of security defense.
End of Support Stage (Stage 4)
Medical devices in Stage 4 are considered more vulnerable than any of the other stages. They may still be in use for providing patient care, but they have been publicly identified as no longer being supported by the medical device manufacturer. Each of these scenarios result in a medical device that cannot be consistently defended against modern cybersecurity dangers.
Critical facets healthcare information technology departments should look for include:
- Medical devices that have been declared EOS by the medical device manufacturer
- Medical devices that are not actively marketed or sold by the medical device manufacturer
- Medical devices that contain software, firmware, or programmable hardware components no longer supported by software developers
- Medical devices with known risks to device safety and effectiveness that are unmitigated
Risk Management
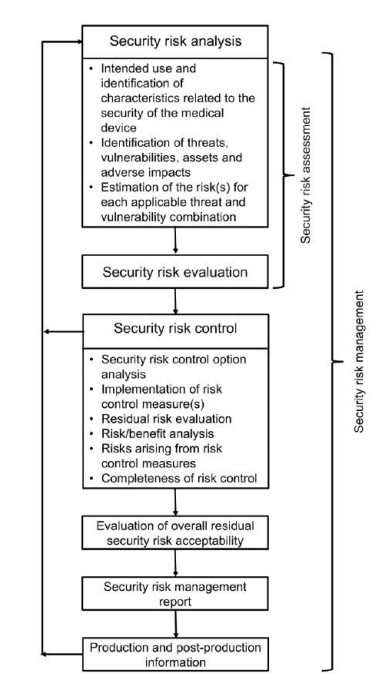 Further, the guidance calls for a risk management approach to product lifecycle management featuring:
Further, the guidance calls for a risk management approach to product lifecycle management featuring:
- Security risk analysis
- Security risk evaluation
- Security risk control
- Security risk acceptability
The cybersecurity guidance expands on product design and how security is incorporated and maintained through the product life cycle. This can be accomplished through using risk control and a secure development framework.
Risk mitigation recommendations for medical device manufacturers include:
- Security design and controls based on intended use of the medical device
- Security risk assessments across the risk management process
- Threat modelling to help determine operational risk
Security testing and communication for medical device manufacturers include:
- Customer facing product security documentation & communication
- Post-market monitoring of cybersecurity vulnerabilities
- Identification of vulnerabilities in third party risk management
- Vulnerability risk identification based on the device security design, controls, and mitigations
Ensuring availability of security patches & mitigations based on device risk:
- Coordinated and clear communication to all affected users
- Description related to the vulnerability and its corresponding mitigations
- Identification of other mitigation options when a security patch is unavailable
Data Integrity
One of the core principles the guidance stresses is cybersecurity information, data integrity and the importance of information sharing. IMDRF encourages medical device industry stakeholders to implement a proactive pre- and post-market approach to cybersecurity information sharing.
Moreover, timely information can help the industry recognize threats, evaluate associated risks, and react quickly as needed. An increase in industry transparency could directly benefit healthcare providers, medical device users and medical device companies.
Security Updates
An important section of the medical device cybersecurity guidance details stakeholder responsibilities related communications, risk management, and transfer of responsibility. Specifically, it is important that medical device manufacturer communications are comprehensive & identify types of documentation needed and when the medical device user may need it.
Product Security Documentation
Medical device manufacturers should ideally provide PLC documentation about security or support changes early in the Support stage. This helps HCP risk management during both the procurement & deployment of medical devices. Types of life cycle support for product security documentation includes:
- Manufacturer disclosure statement for medical device security
- Software Bill of Materials (SBOM)
- Security test report summaries
- Third-party security certifications
- Customer security documentation
Product Life Cycle Documentation
Medical device companies should communicate the strategic life cycle milestones to their customers. Further, these interactions would include cybersecurity EOL and EOS dates if available. This helps to support HCPs during both the procurement & installation process.
Additionally, medical device manufacturers should provide this information as far in advance as possible. The goal is at least 2 years in advance to best support healthcare professionals with the following information:
- Affected medical devices
- Medical device operating system(s)
- Version of medical device deployed
- Medical device software components
- Expected date of medical device service changes
- Extent of medical device maintenance after a service change occurs
- Additional design controls that help all involves parties
Vulnerability & Patching Information
If a vulnerability is uncovered, medical device companies should provide related vulnerability information. Further, the guidance specifically mentions the importance of both the appropriate mitigation or available software patch. Additionally, the guidance stresses an elevated priority be placed on high-risk vulnerabilities where timely communication is required. This communication is designed to help prevent both patient injury or device interruption.
Finally, the mitigation method and implementation instructions should be provided to the medical device operators. These security updates include both an over-air update or deployment of service personnel to help install the remedy.
Proactive Communications for Third-Party Components
Medical device software and other digital components within a medical device will reach EOL/EOS before the product itself does. In these cases, risk can increase based on the lack of support for these elements. To help compensate for these security risks, the cybersecurity guidance suggests medical device companies should:
- Validate the list of third-party components used in medical devices
- Track support status updates of third-party components used within their device
- Assess the risks that exist when third-party components become unsupported
- Communicate new risks and available risk mitigations to healthcare providers
To begin the Regulatory Compliance Associates scoping process today, please enter your information in the blue form below and click the submit button at the bottom of the webpage. You may also email us at [email protected].

