
GMPs for Early Stage Development Projects
Share Our Information
GMPs should be in place during the later stages of clinical development where the final safety and efficacy of a product are being established.
Below is the proposal for applying GMPs for Early Stage development projects. The information below is consistent with FDA’s proposals on a “graded” approach in developing and building scientific information to support clinical investigations and industry norms for development activities.
GMPs for Early Stage Material Controls
Non-clinical: All materials used for non-clinical safety testing (GLP) must have adequate documentation of the methods of synthesis and must be characterized for identity, strength, purity and composition using scientifically sound methods. No regulations for GLP testing require GMP materials nor are there any specified minimums regarding the level of “validation” required for methods.
There is a high probability of “process changes” in the method of synthesis at this stage of development; but maintaining the continuity of impurity profiles is important to be able to reference the data generated in the future. Changes can be made, but a determination will be needed to evaluate if existing data will continue to support the new material or if new studies will be required.
Have questions about your GMPs? Contact Us Now →
Methods are generally expected to be stability indicating and must provide a means to verify identity, strength, purity and quality. Defining the impurity profile with sufficient sensitivity is necessary to qualify impurity levels in support of future development and registration activities.
At the IND-enabling-study stages, the key / critical quality attributes will always include purity and impurities
GMPs for Early Stage Quality Specifications
With respect to quality specifications, critical quality attributes shall be monitored, but there is no regulatory expectation of “limits”; a few exceptions are genotoxic impurities and class I / II solvents. At the IND-enabling-study stages, the key/critical quality attributes will always include purity and impurities (organic, inorganic, residual reagents, and solvents, etc.). For injectable products, sterility and bioburden tests are always required. Other attributes that affect bioavailability could be important depending on the dosage form and route of administration.
Specifications, per se, are not required for GLP materials, but test articles are expected to be “acceptable” to meet study needs and appropriately characterized. There are no default limits established, so any material that is used in a GLP test would be acceptable. Business risks become the driver, e.g., too pure or too impure could lead to difficulties later in development or to premature demise of the compound.
Phase I / II Clinical

Specifications (tests, test methods and acceptance criteria) are required starting at Phase I although the use of “report results” for the acceptance criteria is acceptable for some determinations, particularly those attributes that will be defined more by process control rather than established as a means of assuring safety.
Key / Critical quality attributes are defined as those attributes that could have adverse impact on safety or efficacy. Key / Critical quality attributes (such as purity / impurity) will require defined specification limits that are supported by toxicological study data. One recommendation would be to have “Control Specifications” and “Characterization Specifications”.
GMPs for Early Stage Clinical Development
As more clinical development information about the product is known and the manufacturing process is improved, new specifications may be added. Additionally, specifications may move from one specification list to the other or may be eliminated with appropriate justification. Since IND-enabling toxicology studies will continue to support human dosing, process changes to the API synthesis or formulation changes to the drug product must be evaluated against previous impurity profiles of previous GLP lots.
If the change results in a different impurity profile, new toxicology testing might be warranted. So, it is essential that changes be carefully considered before implementation.
GMPs for Early Stage Acute Toxicity
Key/Critical quality attributes (such as purity/impurity) will require defined specification limits that are supported by toxicological study data. Test methods should also have an appropriate level of qualification (validation) during Phase I / II. Minimum standards for method validation and system suitability requirements are attached in Tables II and III. Deviations from these standards should be justified.
Impurity levels at each stage need to be defined, justified and supported by the impurity test method associated with the material. Impurity levels outside of ICH guidelines must include a toxicological justification, as well as appropriate manufacturing controls to limit this impurity or justification as to why manufacturing controls cannot limit the impurity.
Phase III Clinical
In order to advance to Phase III, an investigational product must demonstrate safety and efficacy on a small scale. To support the Phase III program, the materials used for studies should approximate the expected commercial presentation (with allowances for appropriate blinding requirements). This means that the API manufacturing process, solid state properties, dosage form, strengths, manufacturing process, the container-closure system, etc. will be better defined.
GMPs for Early Stage Clinical Pharmacology
As your clinical pharmacology discovery evolves into a commercial product launch, the proposed manufacturing site may also be selected by this time. Ideally, some Phase III supplies will be produced in the commercial facility. By this stage of the program, several aspects of the product should be controlled in a manner similar to commercial GMP controls. It is still possible that some aspects remain undefined (e.g., optimization controls), but all major aspects (quality controls) should be defined to avoid future delays.
Specifications should be established that are similar to the expected commercial specifications where possible. This means that specifications should be set taking into account the safety limits, the process capabilities and the stability characteristics of the product. This is the appropriate time to evaluate and justify every specification. Sometimes companies inadvertently retain legacy specifications that are left over from early development even though they have become irrelevant and should be removed.\
GMPs for Early Stage Standard Operating Procedures
Methods need to be more rigorously validated and should meet ICH standards. Certainly by the time the registration stability studies are initiated, these methods need to be ICH compliant. If the final GMP testing facility is different, appropriate method transfer, revalidation or verification activities shall be carried out and documented appropriately.
Process and analytical change control needs to be strictly enforced at this stage to assure justification of the commercial process is adequate during the pre-approval process.
ICH stability studies will be initiated on the API and drug product. Depending on company’s global registration / filing strategy, it is important to consider all required temperature and humidity conditions. Firms must seriously consider method changes in the middle of on-going stability studies. Showing continued correlation of data at any given time is important to demonstrate the stability.
GMPs for Early Stage Batch Release
Retention samples are required for API and bulk dosage forms for development projects. An amount sufficient to perform the release testing twice (without the sterility and pyrogen testing) is required. This is required for both GLP and GMP manufacturing, but retention samples do not need to be designated as GLP or GMP; therefore, a single sample per lot is acceptable.
Retention samples are also required for packaged clinical supplies. A common practice is to retain one example of a patient supply per label. Depending on the complexity of the study this could mean, for example, one patient kit per study arm per visit.
There are specific requirements for bioequivalence / bioavailability (BA / BE) studies where retention samples are also kept at the site of the study to assure a completely unbiased conduct. When such a situation is approached, consult with QA and the current regulations to assure that the planned study will meet all requirements.
GMPs for Early Stage Facility and Equipment Controls
Clinical supplies do not have to be manufactured and tested (and often are not) at FDA inspected and approved facilities (but if a facility were to be inspected by the FDA, it should be able to pass the inspection without critical deficiencies). This means that major equipment and supporting utilities need to be maintained in a state of control. Calibration and maintenance programs will need to be in place and documentation must be available to support adequate operation.
In some cases, it is feasible to perform the manufacturing at lab scale (although usually not for sterile products) with the use of disposable labware of adequately controlled materials. The IND process allows for flexibility of the production process.
GMPs for Early Stage Production and Process Controls
Non-Clinical
Testing of test articles / materials will need to meet GLP requirements to avoid a GLP Compliance Statement exception in a study report. This includes, but is not limited to, adequate programs to cover training, calibration and maintenance, documentation practices, material controls, Quality Assurance Unit (QAU) review and oversight and data integrity. This may also include computer validation. It is not required nor expected that the release testing of GLP test articles should be done in a GMP lab. Good science and good practices that are defendable are keys.
Phase I / II Clinical
Validation of manufacturing processes is a requirement of the current Good Manufacturing Practice (cGMP) regulations for finished pharmaceuticals and is considered an enforceable element of current good manufacturing practice for active pharmaceutical ingredients (APIs). A validated manufacturing process has a high level of scientific assurance that it will reliably produce acceptable product. The proof of validation is obtained through rational experimental design and the evaluation of data, preferably beginning from the process development phase and continuing through the commercial production phase.
A validated manufacturing process has a high level of scientific assurance that it will reliably produce acceptable product.
Reviews would fall outside of routine production A validated manufacturing process has a high level of scientific assurance that it will reliably produce acceptable product. Four operations and would be conducted to assess procedures, practices, and information including data generated from production and investigational new drug testing. Based on the review, appropriate modifications and corrective actions can be taken to control procedures and production operations.
GMPs for Early Stage Process Validation
Process validation for clinical supplies in Phase I and II requires assuring intra-batch consistency. When producing multiple batches of the same investigational product, it is recommended that internal performance reviews be conducted and documented periodically. It is also recommended that such reviews assess the control and consistency of the production process and overall product quality.
The data generated with each batch can also allow the establishment and / or refinement of acceptance criteria as experience and knowledge permits. This allows the firm to achieve more consistent investigational new drug production. Activities and decisions will be documented and will be the basis for future change management.
Several elements will need to be considered including, but not limited to:
- Identification and characterization of raw materials
- Calibrated and maintained equipment
- Appropriate methods (validated for stage of development)
- Change management
- Independent QA with approval of
- Master production and labeling records
- Production and labeling records
- Specifications (appropriate to stage of development)
- Cleaning strategy (to avoid cross-contamination)
- Process validation (to assure intra-batch consistency – not batch to batch consistency)
- Bioburden and Sterility Assurance for parenterals (flexible only based on batch sizes allowances)
API and drug product stability need to support the use of clinical supplies for the intended duration of the study. Stability studies that meet full ICH guidelines are not required, but the general scientific principles of the ICH Stability Guidance should be followed. For the reasons of timing and expediency, it is acceptable to refrigerate the product initially. If there are stability issues with the API or dosage form, these should be known by the time the IND is filed. The controls applied should be proportional to the stability of your API / Product. Less stable compounds will require more scrutiny, protection, monitoring and controls.
API and drug product stability need to support the use of clinical supplies for the intended duration of the study.
Phase III Clinical
For Phase III, consistency of material quality of the clinical trial materials (CTM) compared to the eventual marketed product is essential. Process validation for commercial product for the US is not required at pre-NDA stage (although protocols are; however, if speed to launch is important, then early validation would be appropriate.
Technology transfer and validation / verification at the final commercial manufacturing site will be required in time to ensure that the associated site / scale / equipment changes have no adverse impact on the quality of the API / drug product. Defining the final primary container closure is important to study the stability of the dosage form representing final commercial product.
A Change Control process (rather than change management) will need to be initiated subsequent to validation activities (e.g., methods) and manufacture of the registration stability lots.
GMPs for Early Stage Records, Documents and Change Control
Non-clinical
Documentation of the manufacturing activities may be notebook based and does not require any pre-approval from QA to meet compliance requirements.
Forced degradation studies shall be conducted to define the “stability indicating” nature of test methods. Identification of major degradants / impurities should be done to the “best possible” extent. Impurities / degradants having known “structural alerts” with major toxicity potential (e.g., DNA intercalators, aldehydes and alkylating agents like halides) should be monitored closely and controlled. As analytical techniques have improved, low levels of genotoxic impurities have become a major focus. They have to be controlled at very low levels (ppm). Also, leachables and extractables should be kept in mind when developing analytical methods and studying the stability data.
Phase I Clinical
For solid oral products the possibility of lab-based manufacturing operations is possible as long as scientific integrity is maintained and the risk associated with the product is not significantly greater than the risk of the new product itself (e.g., the possibility of cross contamination is significantly minimized). The controls listed above in “Production and Process Controls” section will need to be adjusted to cover such operations. Batch records will be required and QA pre and post approval will be necessary. The batch records at this stage may be constructed in a less prescriptive manner with high level steps. In this case, specific details are recorded during the manufacture to allow reconstruction and evaluation of the manufacturing activities.
Sophisticated dose formulations need not be developed in the early phases as long as the scientific information that is obtained from a particular study design can allow movement to the next step. Phase I supplies are often only API without excipients.
Phase II / III Clinical
Process development activities need to be thoroughly documented at this stage of development. Much of the process control information will be maintained and presented in development reports which need to be available for a Pre-Approval inspection. This will also be the stage where critical process controls will be documented and made available to the commercial manufacturing site through a Technology Transfer process.
Changes to processes and methods used for the clinical Phase III materials will need to be fully documented and justified since they will be a reference point for the commercial processes during a Pre-Approval inspection.
GMPs for Early Stage Regulatory Requirements
Non-Clinical
Information to support Pre-IND briefing documents will follow GLP guidelines listed in Table I. The extent of required documentation will vary and be dependent upon the nature and complexity of the investigational drug.
IND Submission and Phase I through III
Information submitted must demonstrate that all aspects of manufacturing and controls are performed under cGMP as defined in Table I.
The CMC section should discuss the composition, manufacture and control of the API and dosage form (DP) as appropriate for the particular investigations covered in the IND. Sufficient information should be provided to assure the proper identity, quality, purity and strength of the investigational drug.
The FDA recognizes that modifications to the method of preparation of the API and DP and changes in the DP itself are likely as the investigation progresses. The emphasis in an initial Phase I submission should generally be placed on the identification and control of the raw materials and the new drug substance.
The amount of information submitted will depend on the scope of the clinical investigation and will vary with each phase and duration of the investigation, the dosage form and the information otherwise available. The sponsor should submit information amendments to supplement the original information submitted as the drug development proceeds and as the scale or production changes from pilot scale production appropriate for the limited initial investigations to the larger scale production needed for expanded clinical trials.
Reflecting the distinctions described above and based on the phase(s) to be studied, the submission is required to contain the following:
(a) API:
- A description of the drug substance, including its physical, chemical or biological characteristics
- Name and address of the API manufacturer
- General method of preparation of API
- Acceptable limits and analytical methods used to assure identity, strength, quality and purity of the API
- Information sufficient to support stability of the API during the toxicological studies and the planned clinical studies
- Brief and general description of the composition, manufacture and control of any placebo used in a controlled clinical trial
NOTE: References to current USP/NF may satisfy relevant requirements
(b) Drug Product:
- List of all components used in the manufacture of the DP (this may include reasonable alternatives for inactive components) including those components intended to appear in the DP and those which may not appear, but which are used in the manufacturing process.
- Where applicable, the quantitative composition of the investigational DP
- Name and address of the DP manufacturer
- Brief general description of the manufacturing and packaging of the DP 6
- Acceptable limits and analytical methods used to assure identity, strength, quality and purity of the DP
- Information sufficient to assure the stability of the DP over the course of the study
NOTE: References to current USP / NF may satisfy relevant requirements
The amount and level of detail for the information to support the CMC should continue to expand as more information about the DS and DP composition, its methods of manufacture and packaging, analytical methods and specifications and stability data become more defined.
Table 1. Pre-IND Briefing Requirements

Notes:
- Phase IV is not included in this table
- Assumptions for this presentation include:
-
- Timelines that would allow the decisions on the commercial presentation and process prior to, or early in Phase III.
- Selection of commercial suppliers and CMOs prior to or early in Phase III.
- Product characteristics are “standard”. The existence of “non-standard” characteristics such as identified sensitivities may require adjusting information and controls forward or back.
-
Table II. Method Performance, Validation, and Documentation Requirements per Phase of Development – Minimum Standards
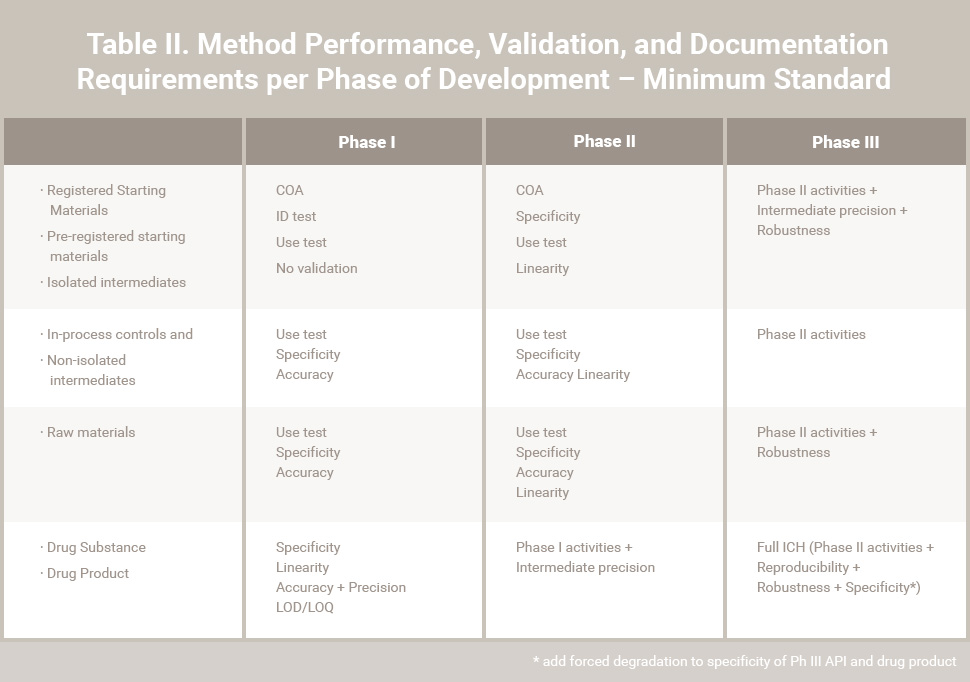
Table III. System Suitability Requirements per Method – Minimum Standards

About RCA’s Pharmaceutical Consulting Services
Regulatory Compliance Associates (RCA) has helped thousands of pharmaceutical companies meet regulatory, compliance, quality assurance, and remediation challenges. With more than 20 years of experience with FDA, Health Canada, EU and global regulatory agencies worldwide, Regulatory Compliance Associates® offers leading pharmaceutical consultants. We’re one of the few pharma consulting companies that can help you navigate the challenges associated with industry regulations.
Our pharmaceutical consulting firm includes over 500 seasoned FDA, Health Canada & EU compliance consultants and regulatory affairs experts who understand industry complexities. It’s a pharma consultancy founded by regulatory compliance executives from the pharmaceutical industry. Every pharmaceutical industry consultant on the Regulatory Compliance Associates team knows the unique inner workings of the regulatory process.
Client Solutions
Whether you’re in the product planning, development or pharmaceutical lifecycle management stage or need a remediation strategy for a compliance crisis, Regulatory Compliance Associates will guide you through every pharmaceutical consulting step of the regulatory process and create a customized approach depending on your product and your pharma company’s individual needs. Our regulatory compliance clients include:
- Companies new to FDA, Health Canada or EU regulations and regulatory compliance
- Start-up organizations with novel submissions to 510(k) submissions from multi-national corporations
- Investment firms seeking private equity due diligence for pre-acquisition and post-deal research
- Law firms seeking pharmaceutical consulting firm expertise in the remediation of warning letters, consent decrees, 483’s or import bans
Regulatory Affairs
Regulatory affairs is Regulatory Compliance Associates backbone. We exceed other pharma consulting companies with industry experts experienced in complexities of the pharmaceutical and biopharmaceutical industries. Our pharma consulting expertise spans all facets and levels of Regulatory Affairs, from Regulatory Support for New Products to Life Cycle Management, to other services like Outsourced Regulatory Affairs, Submissions, Training, and more.
As your partner, we can negotiate the potential assessment minefield of regulatory compliance services with insight, hindsight, and the clear advantage of our breadth and depth of knowledge and regulatory compliance consulting. We offer the following pharma consulting regulatory affairs services for pharmaceutical companies.
- New Product Support
- Product Lifecycle
- Other Regulatory Services
- Combination Products
Compliance Assurance
The regulations process surrounding pharmaceutical companies can be tricky for even the most experienced industry veteran to understand. Just one misstep could mean significant and lasting consequences for your business. At Regulatory Compliance Associates, we offer the pharma consulting experience and pharma consultants necessary to guide you through the quality compliance process.
- Assessments
- Audits
- Regulatory Agency Response
- Preparation and Training
- Inspection Readiness
- Data Integrity
Quality Assurance
Regulatory Compliance Associates Quality consulting includes assessments, strategy, implementations, staff augmentations, and identification of quality metrics to ensure continuous improvement. Our pharma consultants understand the strategic thinking needed to align your business needs and goals. Regulatory Compliance Associates quality assurance services include quality experts with experience spanning major corporations and start-ups. Our pharmaceutical consulting firm knows firsthand how to achieve, maintain, and improve quality, and we excel in transferring pharma consulting knowledge to your organization.
- 21 CFR Part 11
- Data Integrity
- Manufacturing Support
- Facility Support
- Quality Metrics
Remediation Services
Regulatory Compliance Associates has a proven remediation services approach to managing FDA Warning Letters, Consent Decrees, Remediation and other serious regulatory situations. Our pharma consultants know how to partner with executive, legal, and communication teams. Each RCA pharma consulting Expert will develop a response that will be accepted by the regulatory agency and be realistic to execute.
Regulatory Compliance Associates pharma regulatory consultants will develop a comprehensive proof book of documented evidence demonstrating the corrective action taken to remediate non-compliant issues. In addition, each Regulatory Compliance Associates pharma consulting Expert understands compliance enforcement. We’ll prepare a comprehensive pharma consulting strategy to assist in your remediation efforts, drive continuous improvement, and maintain regulatory compliance with the regulations.
- Regulatory Action
- Regulatory Compliance
- Regulatory Enforcement
- Warning Letter
- 483 Observation
- Oversight Services
- Risk Management Plan
About Regulatory Compliance Associates
 Regulatory Compliance Associates® (RCA) provides pharmaceutical consulting to the following industries for resolution of life science challenges:
Regulatory Compliance Associates® (RCA) provides pharmaceutical consulting to the following industries for resolution of life science challenges:
- Life Sciences
- Pharmaceutical
- Biologic & Biotechnology
- Sterile compounding
- Medical device
- Lab Testing
We understand the complexities of running a life science business and possess areas of expertise that include every facet of R&D, operations, regulatory affairs, quality, and manufacturing. We are used to working on the front lines and thriving in the scrutiny of FDA, Health Canada, MHRA and globally-regulated companies.
As your partners, we can negotiate the potential minefield of regulatory compliance and regulatory due diligence with insight, hindsight, and the clear advantage of our unique expertise and experience.
- Founded in 2000
- Headquartered in Wisconsin (USA)
- Expertise backed by over 500 industry subject matter experts
- Acquired by Sotera Health in 2021
About Sotera Health
The name Sotera Health was inspired by Soteria, the Greek goddess of safety, and reflects the Company’s unwavering commitment to its mission, Safeguarding Global Health®.
Sotera Health Company, along with its three best-in-class businesses – Sterigenics®, Nordion® and Nelson Labs®, is a leading global provider of mission-critical end-to-end sterilization solutions and lab testing and advisory services for the healthcare industry. With a combined tenure across our businesses of nearly 200 years and our industry-recognized scientific and technological expertise, we help to ensure the safety of over 190 million patients and healthcare practitioners around the world every year.
We are a trusted partner to 5,800+ customers in over 50 countries, including 40 of the top 50 medical device companies and 9 of the top 10 pharmaceutical companies.
Commitment to Quality
Our Certificate of Registration demonstrates that our Quality Management System meets the requirements of ISO 9001:2015, an internationally recognized standard of quality.
To begin the Regulatory Compliance Associates scoping process today, please enter your information in the blue form below and click the submit button at the bottom of the webpage.